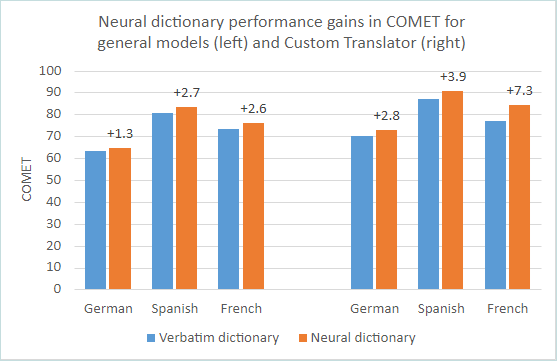
Zespół SAPHO nie jest chorobą, która przebiega według ściśle określonego schematu. Cechuje go współwystępowanie zmian kostno-stawowych, skórnych i narządowych. Nazwa zespołu to akronim od charakterystycznych objawów: Synovitis (zapalenie błony maziowej stawów) - Acne (trądzik) - Pustulosis (łuszczyca krostkowa dłoni i stóp) - Hyperostosis (nadmierny rozrost kości) - Osteitis (zapalenie kości). Zespół SAPHO to schorzenie autoimmunologiczne, do którego dochodzi z nieznanych przyczyn. Na dzień dzisiejszy pozostaje niewyleczalny, ale farmakologicznie można łagodzić jego objawy.
Zespół SAPHO nie jest chorobą, która przebiega według ściśle określonego schematu. Cechuje go współwystępowanie zmian kostno-stawowych, skórnych i narządowych. Nazwa zespołu to akronim od charakterystycznych objawów: Synovitis (zapalenie błony maziowej stawów) - Acne (trądzik) - Pustulosis (łuszczyca krostkowa dłoni i stóp) - Hyperostosis (nadmierny rozrost kości) - Osteitis (zapalenie kości). Zespół SAPHO to schorzenie autoimmunologiczne, do którego dochodzi z nieznanych przyczyn. Na dzień dzisiejszy pozostaje niewyleczalny, ale farmakologicznie można łagodzić jego objawy.Synonimy:
ang. Synovitis-acne-pustulosis-hyperostosis-osteitis syndrome
Rozpowszechnienie: nieznane. Dane są niedoszacowane, ale zespół uznawany jest za chorobę rzadką.
Przyczyny
Niejasne. Choroba autoimmunologiczna, w której rolę może odgrywać czynnik infekcyjny (Chlamydia spp. lub Yersinia spp.). Choroba może występować rodzinnie.
Objawy i diagnostyka
Choroba przebiega z okresami zaostrzeń objawów i remisjami, czyli czasem, w którym "choroba jest uśpiona". Zaostrzenia choroby mogą zostać wywołane przez stres i emocje.
Do najczęstszych objawów kostno-stawowych należą: bóle przedniej ściany klatki piersiowej (najczęściej choroba dotyczy klatki piersiowej), bóle kręgosłupa oraz obrzęki stawów mostkowo-obojczykowych. Rzadziej dochodzi do zajęcia pozostałych stawów.
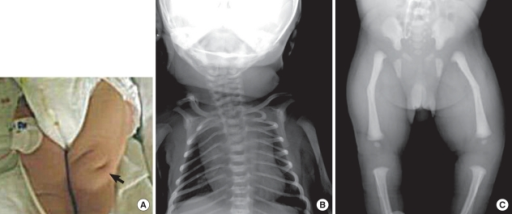
Hipertrofia kości lokalizuje się zwłaszcza w obrębie kręgosłupa, a także miednicy i spojenia łonowego oraz mostka. Mogą także pojawić się osteofity na kręgach oraz pogrubienie warstwy korowej kości długich (kości na zdjęciu rentgenowskim są widoczne jakby "obrysowane" białym konturem).
Zapalenie kości najczęściej jest jałowe, czyli niespowodowane bakteriami (jednak czasem w posiewach materiału kostnego występuje bakteria Propionibacterium acnes). Zwykle występuje w obrębie kości klatki piersiowej, a następnie kręgosłupa, kości długich lub też żuchwy i stawów krzyżowo-biodrowych.
W szpiku kostnym również stwierdza się zmiany (zwiększenie plazmocytów i monocytów, komórek olbrzymich), a w późniejszym etapie choroby zmniejszenie komórek szpikowych i włóknienie szpiku, co będzie wpływało na pojawienie się cytopenii (obniżenie poziomu elementów morfotycznych krwi).
Najczęstszymi zmianami skórnymi w przebiegu zespołu SAPHO, są łuszczyca krostkowa rąk i stóp oraz wypryski o typie trądziku (zwykle na klatce piersiowej i na plecach), choć brak zmian skórnych nie wyklucza rozpoznania choroby. Pojawienie się zmian skórnych w fazie zaostrzenia nie musi być związane z toczącym się zaostrzeniem w obrębie kości i stawów. Zmiany mogą przybierać formę krostek, łuszczących się miejsc oraz pęcherzyków.
Do rozpoznania nie jest konieczne stwierdzenie występujących jednocześnie problemów skórnych i kostnych. Zmiany skórne nie zawsze występują. Mogą też pojawić się dużo wcześniej lub później niż kostne.
Badania diagnostyczne
Badaniami pomocnymi w postawieniu diagnozy są:
- morfologia krwi i badania biochemiczne (wysokie OB, CRP, leukocytoza, nieznaczna niedokrwistość, zwiększenie stężenia fosfatazy alkalicznej); antygen HLA-B27 jest obecny u niewielu chorych; czynnik reumatoidalny i przeciwciała przeciwjądrowe są nieobecne;
- badania mikrobiologiczne - w posiewach materiału kostnego czasem ujawnia się Propionibacterium acnes;
- zdjęcia rentgenowskie (w których zwracają uwagę nadżerki w stawach mostkowo-obojczykowych, sklerotyzacja podchrzęstna (podchrzęstne zagęszczenie struktury kostnej), zapalenie okostnej, zwapnienia więzadeł żebrowo-obojczykowych, zwapnienia okołokręgowe i obecność syndesmofitów na kręgach; gdy choroba trwa długo można stwierdzić sklerotyzację pojedynczych kopści);
- USG stawów, scyntygrafia kości, czasem rezonans magnetyczny.
Kryteria rozpoznania zespołu SAPHO
Do rozpoznania konieczne jest stwierdzenie przynajmniej 1 z 3 poniższych kryteriów:
1) przewlekłe nawracające zapalenie kości i szpiku:
• jałowe (w pojedynczych przypadkach Propioniobacterium acne),
• zajęcie kręgosłupa, przedniej ściany klatki piersiowej – szczególnie częste zajęcie obojczyków i pierwszych żeber,
• ze zmianami lub bez zmian skórnych,
2) zapalenie stawów i współistniejące zmiany skórne, takie jak:
• trądzik (SA) a. conglobata, ulcerans, hydraadenitis suppurativa),
• łuszczyca krostkowa rąk i stóp (PPP),
• łuszczyca zwykła,
3) każde zapalenie kości współistniejące z:
• łuszczycą krostkową rąk i stóp, łuszczycą zwykłą, trądzikiem.
Różnicowanie:
- zesztywniejące zapalenie stawów kręgosłupa,
- łuszczycowe zapalenie stawów,
- szpiczak mnogi,
- mięsak Ewinga,
- nowotworowe zmiany w kościach,
- alkaptonuria,
- DISH (zesztywniająca hiperostoza, choroba Forestiera),
- powikłania kostno-stawowe po leczeniu retinoidami,
- niedoczynność tarczycy,
- keratodermia dłoniowo- podeszwowa,
- choroba Pageta,
- bakteryjne zapalenie kości i szpiku.
Warto wiedzieć, że zespół SAPHO może współwystępować z wrzodziejącym zapaleniem jelita grubego i chorobą Leśniowskiego-Crohna.
Możliwości leczenia
Z uwagi na jeszcze niejasne dla medycyny przyczyny choroby, brakuje skutecznych metod leczenia choroby. Możliwe jest jednak postępowanie objawowe. Powinno być holistyczne, indywidualnie dobrane do stanu i oczekiwań pacjenta.
Początkowo stosowane są niesteroidowe leki przeciwzapalne i przeciwbólowe. Jeśli takie leczenie przestaje być skuteczne wówczas włącza się terapię lekami sterydowymi - doustną lub dożylną. Uzupełniająco lekarz może zlecić sulfasalazynę, gdy zajęte są stawy w obrębie klatki piersiowej, kręgosłupa i krzyżowo-biodrowych. Jeśli proces chorobowy obejmuje stawy obwodowe, wówczas rozważa się leczenie metotreksatem. Stosowane są także antybiotyki - głównie makrolidy.
Lekami wspomagającymi terapię są preparaty kalcytoniny i bisfosfoniany, jak pamidronian i zoledronian. Zapobiejgają niszczeniu kości, a także uzupełniają terapię przeciwbólową. Pamidronian i zoledronian działają też przeciwzapalnie.
Gdy powyższe metody leczenia zawodzą, wtedy może zostać wdrożona terapia biologiczna inhibitorami czynnika martwicy nowotworów TNF-alfa (Etanercept). Proponowane jest także leczenie azytromycyną. Należy pamietać, że w przypadku ozdrowieńców po gruźlicy istnieje ryzyko ponownego zachorowania po terapii TNF-alfa. Jest to jednak zjawisko rzadkie.
W leczeniu trądziku stosowane są antybiotyki jak np. doksycyklina oraz izotretynoina (stereoizomer kwasu all-trans-retinowego), zmniejszająca aktywność gruczołów łojowych oraz ich sprzyjająca ich inwolucji (powrót do wielkości naturalnej). Izotretynoina działa również słabo przeciwzapalnie i przeciwnowotworowo. Łuszczyca krostkowa na ogół jest łagodzona kortykosterydami, a także fotochemioterapią (PUVA).
Oprócz metod leczenia ukierunkowanych na objawy zespołu SAPHO, w razie potrzeb wdrażana jest psychoterapia, leki przeciwdepresyjne i przeciwlękowe, gdyż u pacjentów tych często występuje depresja. Ze wspomnianymi wyżej terapiami, jak również specyfiką zespołu wiąże się możliwość wystąpienia cech zespołu zmęczenia chorobą przewlekłą, na co również powinien zwrócić uwagę lekarz prowadzący. Istotną rolę w poprawie komfortu życia pacjenta odgrywa skuteczna terapia bólu. Ponadto u chorych zalecana jest fizjoterapia.
Niestety choroba postępuje w czasie. Jej powikłania, do których zaliczamy zwiększenie podatności kości na złamania, ograniczenie funkcji stawów, ucisk na naczynia, przewlekły ból oraz spondyloartretyzm, negatywnie wpływają na jakość życia pacjentów.
Opracowano na podstawie:
- Maślińska M. Zespół SAPHO – opis przypadku. Reumatologia 2005; 43, 4: 222–226
- Gerkowicz A. et all. Zespół SAPHO jako wyzwanie terapeutyczne – opis przypadku. Przegl Dermatol 2011, 98, 400–404
- Zielińska A. et all. Zespół SAPHO – odmienność przebiegu – trudności diagnostyczne. RU 2006; 44, 4: 213-219
- SAPHO Syndrome. Orphanet. 2014
- Zespół SAPHO. Reumatologia24.pl. Dostęp z dn. 6.10.2015 r.